
Computational prediction of material properties from atomic to microstructural scales is a key, cross-cutting strength of the MSE department at Rensselaer. It provides a framework for understanding the detailed role of individual parameters such as composition, surface structure and chemistry, microstructure, nature of defects and their distribution in material synthesis, processing and properties. Expertise in our department spans electronic structure calculations at the atomic-nano scales, molecular dynamics simulations at the nano-meso scales, phase field simulations at the meso-micro scales, and continuum simulations up to the macroscopic scale. These techniques facilitate in silico prediction of a vast array of properties including chemical, mechanical, phase equilibria and transport of charge, mass and heat, in materials of all classes.
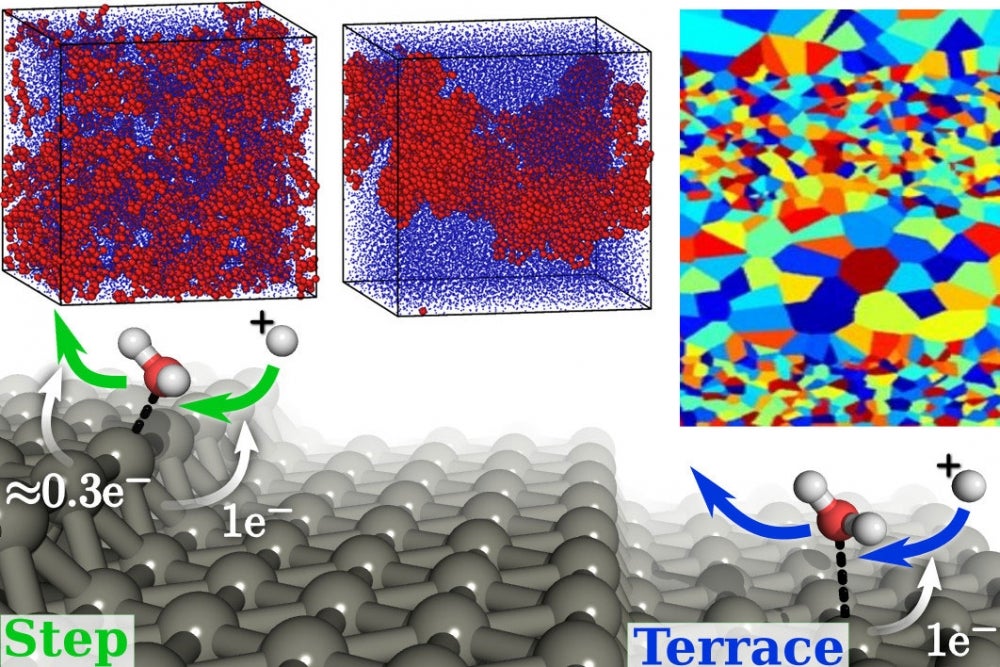
Specific current areas of research, starting at the atomic scale, include first-principles electronic structure calculations of charge transport in nanoscale wires for future electronic devices, and dynamics of hot photo-excited carriers in metallic nanostructures for novel solar cells and photodetectors. Electronic structure calculations also guide the design of electrochemical interfaces for energy conversion and new functional perovskite materials. Molecular dynamics simulations power a vast spectrum of research at Rensselaer including the design of nanostructured materials for energy applications, mass and thermal transport, dynamics of molecular motors, proteins and other biomaterials, properties of polymer nanocomposites, nanofluids, oxide glasses and metallic glasses, as well as interfaces between these materials.
Phase field simulations and diffuse interface models facilitate simulation of meso to micro scale properties of materials, including microstructural evolution, solidification, grain growth, electrochemical transport, ferroelectric switching dynamics and biological membranes. Our department also leads the development of open-source software tools for computational MSE, including the Mesoscale Microstructure Simulation Project (MMSP) and the joint density functional theory software (JDFTx) for multi-scale electronic structure simulations, resulting in a wide-ranging impact on the computational MSE research community as a whole.